

In-Context Learning with Evo2 for RNA Design

Here at Therna we are building programmable RNA therapeutics. We run a Lab in the Loop where our internal models propose RNA sequences, the bench validates them, and the resulting data goes back into the next round of training. The models get refined, and the next design batch gets better.
An in-silico version of this loop leans on two kinds of models doing two different jobs. A generator proposes new sequences, and a predictor scores them. These are usually separate models. The Evo2 paper is a good example, using Evo2 to generate DNA and a predictor like Borzoi to guide it.
I got curious whether in context learning could collapse that split. If I show a foundation model a handful of high activity sequences and ask it for one more, will it return something that also looks high activity? No fine tuning, no numeric labels, just a list of “preferred” sequences in the prompt. If it works, the generator is doing the predictor’s job on its own, inferring “what success looks like” from the examples alone.
To test this I needed two kinds of models.
As the generator I tried two foundation models. Evo2, the long context DNA language model from the Arc Institute, is trained on a broad slice of natural genomes and transcriptomes; I used the seven billion parameter checkpoint. EVA is a recent state of the art RNA foundation model. One is broad and genome scale, the other is RNA specific, which is what makes the comparison interesting.
As the final arbiter of success I used T-Grammar, our proprietary regression model for RNA regulatory activity. Given a 5’ or 3’ UTR, it predicts two things per cell type: activity, how active the sequence is (think expression or stability), and cell specificity, how much that activity differs across cell types. Here T-Grammar is only the judge and is not part of the loop; it scores the generated sequences after the fact and never steers the generation.
The setup
For a single cellular context, I took an in-house dataset measuring the impact of synthetic short variable 5’ UTR sequences on expression and split it into a highly activating set and a highly repressive set based on the experimental measurements. The sequences are fully synthetic and the foundation models have never seen them in training. From each pool, I sampled some number of sequences and concatenated them into a single prompt via a <sep> token.
Then I generated a fixed number of nucleotides at a given temperature, producing a synthetic sequence. I built a bunch of these prompts, half from the activating set and half from the repressive set, generated a batch of sequences per prompt, and passed the sequences through T-Grammar to read out predicted activity and predicted cell-type specificity. Each box and point cloud below summarizes 320 generated sequences per number of examples in the prompt, which matters for reading the significance numbers later.
I also swept the number of in context examples rather than fixing it. For Evo2 I went from two examples in the prompt up to 256. For EVA I went up to 128, which is as far as that model would accommodate. Sweeping the example count is what lets us answer the more interesting question, which is not just whether in context learning happens but how much priming each model needs before the signal becomes clear, and whether activity and specificity behave the same way.
If the in context idea works, the continuations from high prompts should score higher on T-Grammar than the continuations from low prompts. If it does not, the two distributions should look identical. Neither model has any other way to know which prompts came from which pool, because no labels were ever provided.
Evo2:
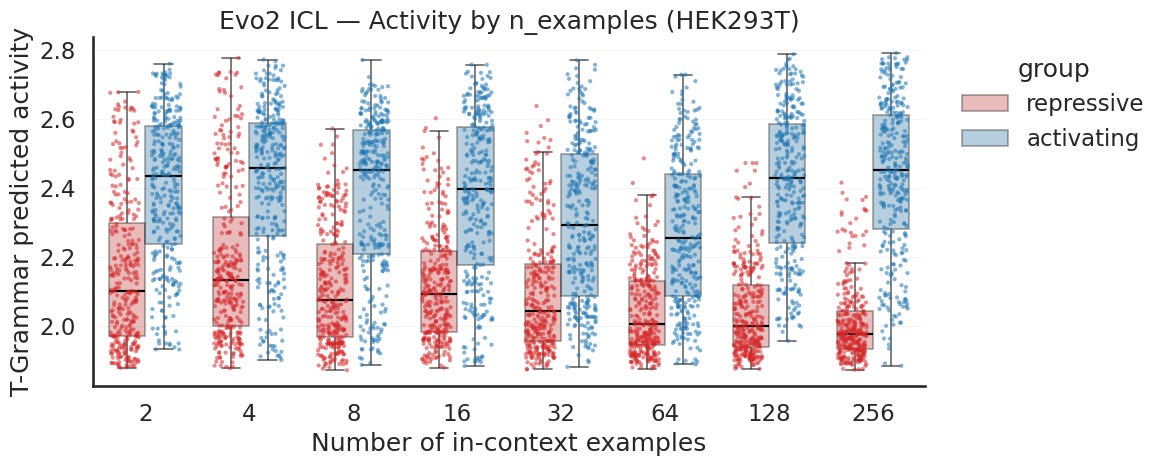
Let’s start with activity. Across every example count from two to 256, the activating primed generations sit above the repressive primed generations. The activating cluster around 2.4 to 2.45 in predicted center of mass and the repressive sit closer to 2.0 to 2.1. The gap is present in two examples and widens as you add more, mostly because the repressive group drifts down while the activating stays put. This is the more legible of the two signals.
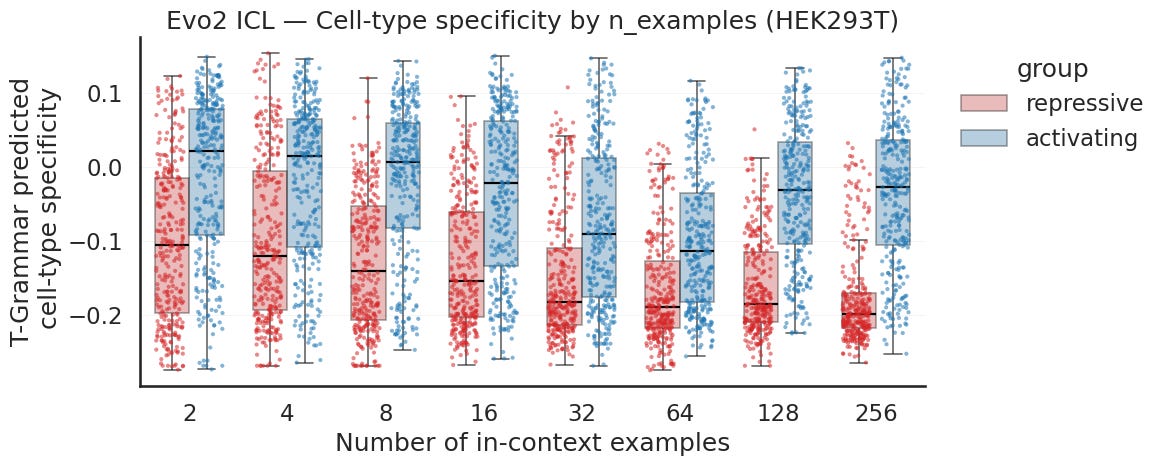
Specificity is the harder readout, and it is a harder biological problem to begin with. The activating primed continuations carry a higher predicted differential signal than the repressive primed ones at every example count, but the two groups sit much closer together than they did for activity. The difference for specificity is also lower in the actual samples used in the prompt. The activating stays near zero, dipping toward the middle of the sweep, while the repressive drifts steadily more negative as you add examples. So the separation is there from the first prompt but it is a smaller gap similar to the input data. Where a sequence pushes activity in one cell type versus another is a finer question than how active it is overall, so it makes sense that it is the harder pattern to pick up.
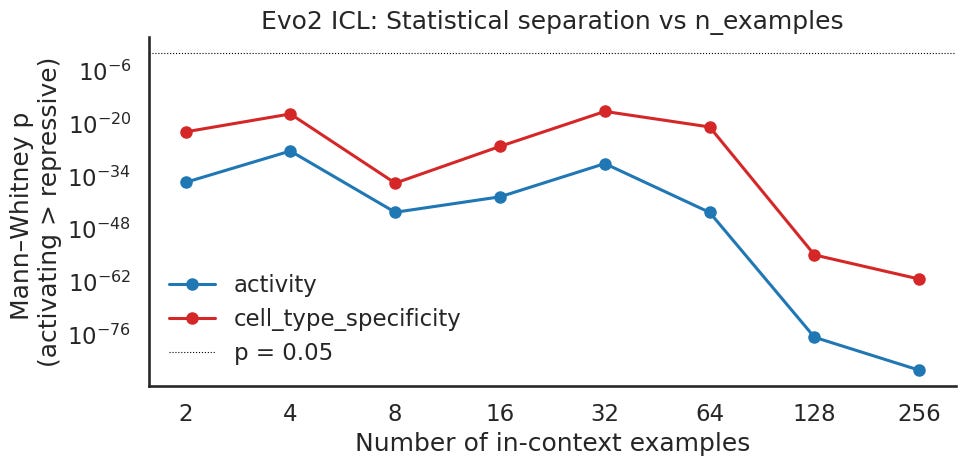
This panel makes the activity versus specificity comparison easier. Two things to notice. First, both signals are significant across the entire sweep by a wide margin, so neither is borderline anywhere, even at the smallest prompt. Activity is the stronger of the two at every count and pulls further ahead at the high end. Specificity runs consistently above activity, less significant but still far below the line the whole way. Both dip a little toward the middle of the sweep before dropping steeply at the top. Second, the message here is the gap between the two curves rather than the absolute values. With a large sample, a very small p value mostly tells you a separation is real, not how large it is. The reading is that specificity is reliably less separated than activity, even though both clear significance easily.
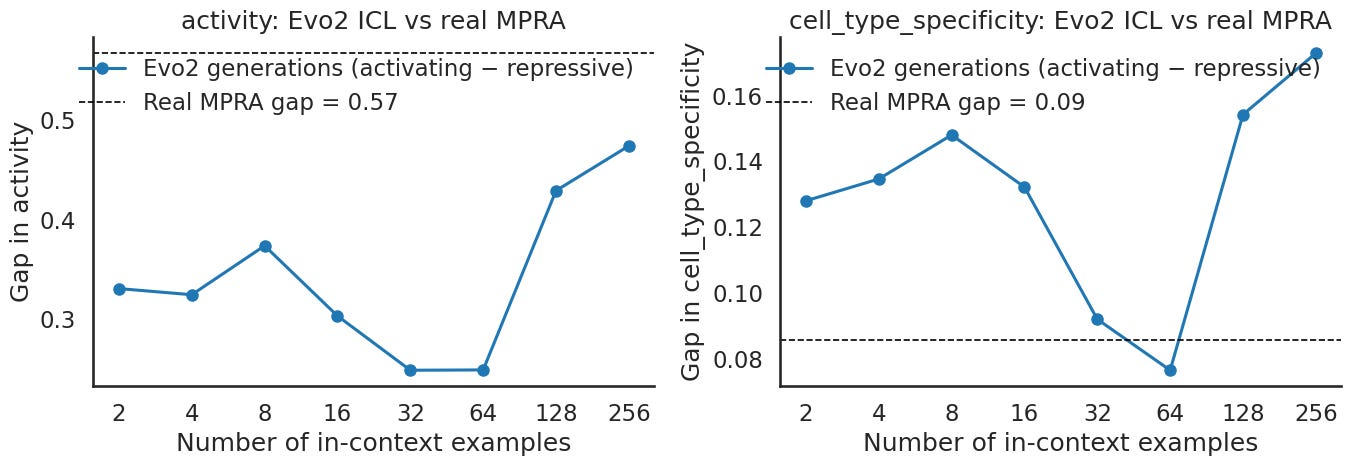
So the last Evo2 panel asks how large the generated gap is in absolute terms, not just whether it is significant. The dashed line marks the gap between the real activating and repressive MPRA pools, the ceiling we are chasing. For activity, the generated gap climbs as you add examples and by the end of the sweep is reaching that ceiling. For specificity, the generated gap sits above its ceiling across almost the whole range. Put another way, Evo2 produces sequences whose predicted activating minus repressive separation is as large as the real pools for activity and larger than the real pools for specificity, and the separation generally grows with more examples.
So the Evo2 takeaway is two parts. Activity is the cleaner, larger signal and shows up from the first prompt. Specificity is real and significant throughout, but it is the smaller and less separated of the two.
EVA:
EVA also does the task, once it is given more than a couple of examples. It takes the same <sep> delimited prompts, it scales up to the same range, and it produces continuations that T-Grammar can score. On the basic question of whether in context learning happens at all, EVA clears it for activity.
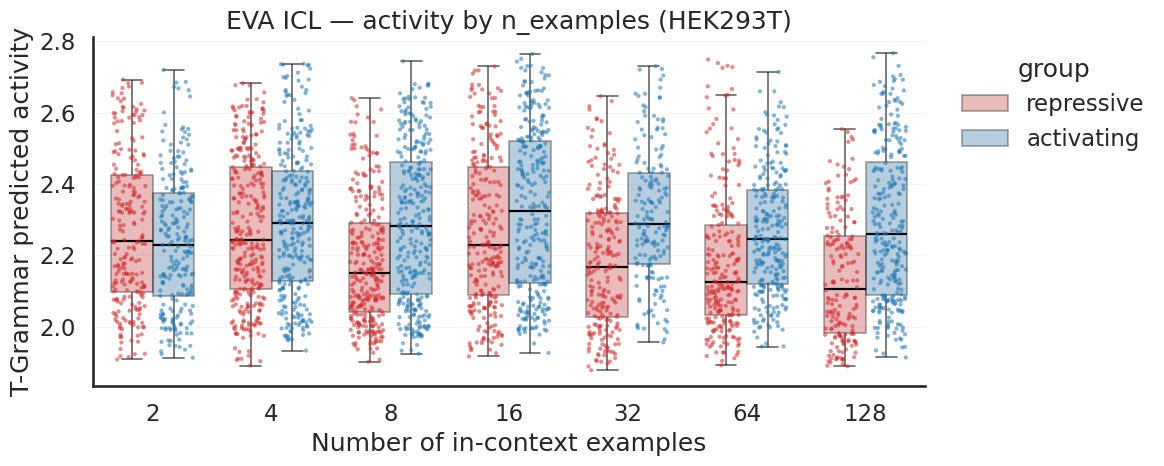
For activity, the activating primed boxes sit above the repressive primed ones across most of the sweep, so the direction is right, with one exception: at the two smallest prompts the groups are essentially tied. After that the activating pulls ahead, and the gap widens as the repressive group drifts down while the activating holds steady. So on the boxes EVA does strengthen with more examples for activity. The separation is still smaller than for Evo2, and as the significance panel shows, it is far less consistent from one count to the next.
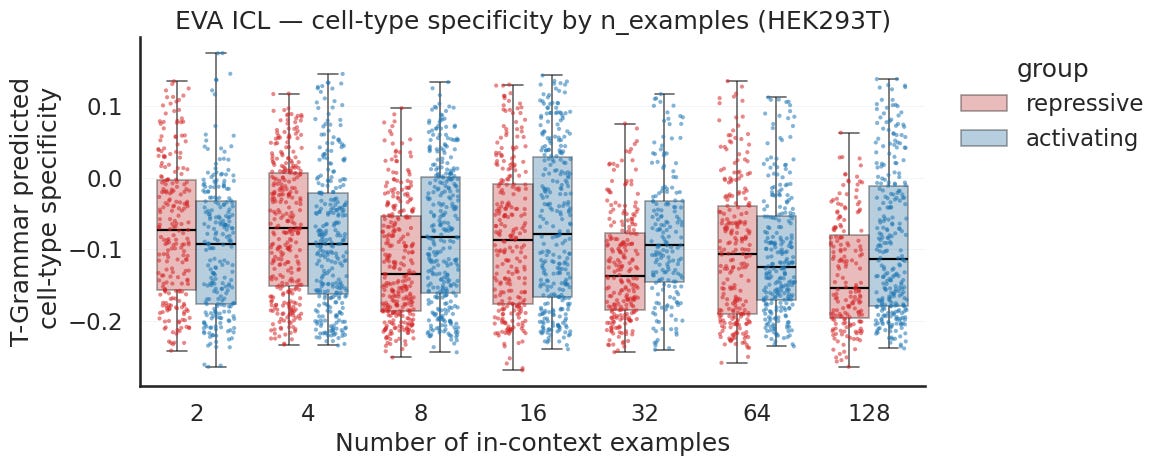
The specificity signal is far fainter and, more tellingly, not consistent in direction. The activating and repressive groups sit close together at most counts, and at times the ordering flips, with the repressive group sitting at or above the activating group. At the counts where it does point the right way, the gap is small. This is the same activity versus specificity pattern as before, except for EVA specificity it is weak enough that even the sign is not stable.
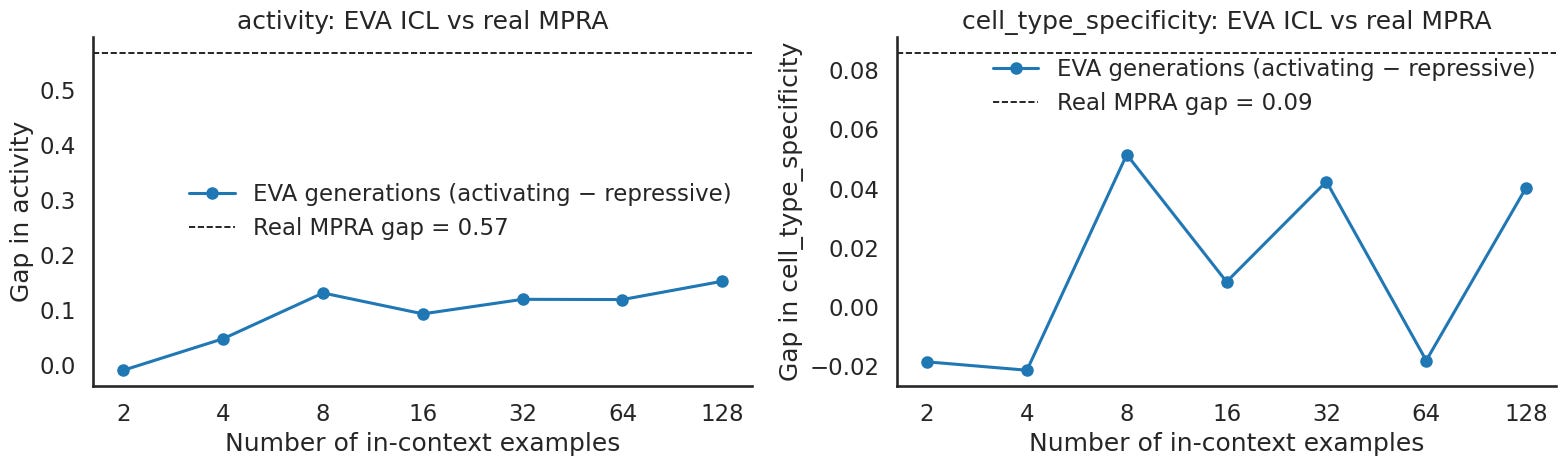
The separation curves tell the same story. Activity separation trends upward as you add examples, while specificity separation stays small and flat. So whatever EVA gains from a longer prompt shows up in activity, not in specificity. That is the opposite of Evo2, where both gaps grew toward and past the real pool ceiling.
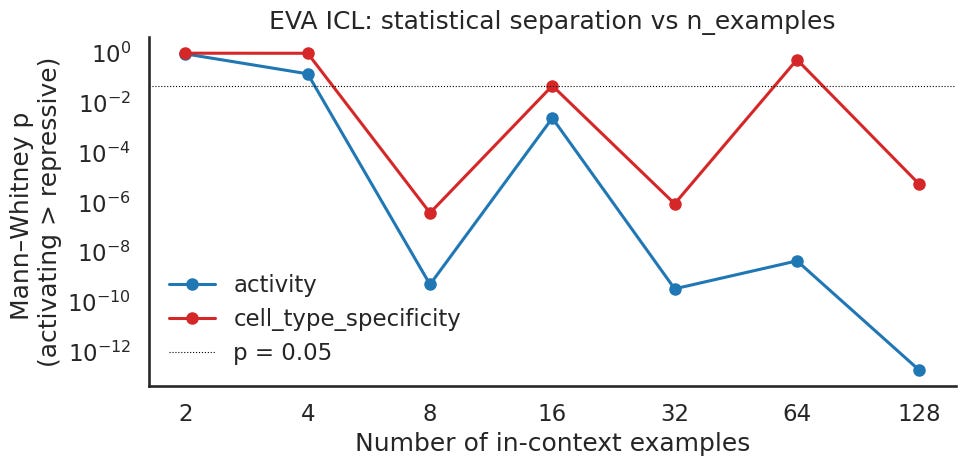
The significance panel is the clearest place to compare the two, and with a large sample it is unforgiving. For EVA, activity is not significant at all at the two smallest prompts. After that it does separate, but along a jagged, sawtooth path: strong at one count, weak at the next, reaching its best only at the high end, where it finally rivals Evo2. Specificity is worse. It fails the significance line at the smallest prompts and again partway through the sweep, lands right on the threshold at one count, and clears it cleanly at only a few. There is no smooth trend, and at several counts the specificity split is not statistically real. Evo2, by contrast, was significant at every count by a wide margin.
Putting the two side by side, the contrast is sharp. Evo2 separates activating from repressive at every example count, for both readouts, by a wide margin. EVA separates cleanly only at some counts, shows no separation at the smallest prompts, and on specificity is inconsistent enough that the direction sometimes flips. The larger sample is part of why this is now visible, since an earlier, smaller run made EVA look more uniformly significant than it is. I would still be careful about reading too much into why. The two models are different in kind. Evo2 is a broad genome scale model and EVA is RNA specific, the two were run over different ranges of example counts, and this is all in silico. One reasonable hypothesis is that the wider training grammar behind Evo2 carries into the fine structure function relationship that specificity depends on, but that is a guess, and the cleaner test is the wet lab rather than more in silico comparison.
Are the generations copies of the prompt?
One worry hangs over all of this. If the model just parrots the sequences in the prompt, the steering is not steering at all, it is the prompt played back, and the effect would only look stronger as you add more examples. So for every generated sequence I measured its highest identity to any of the in context examples it was shown.
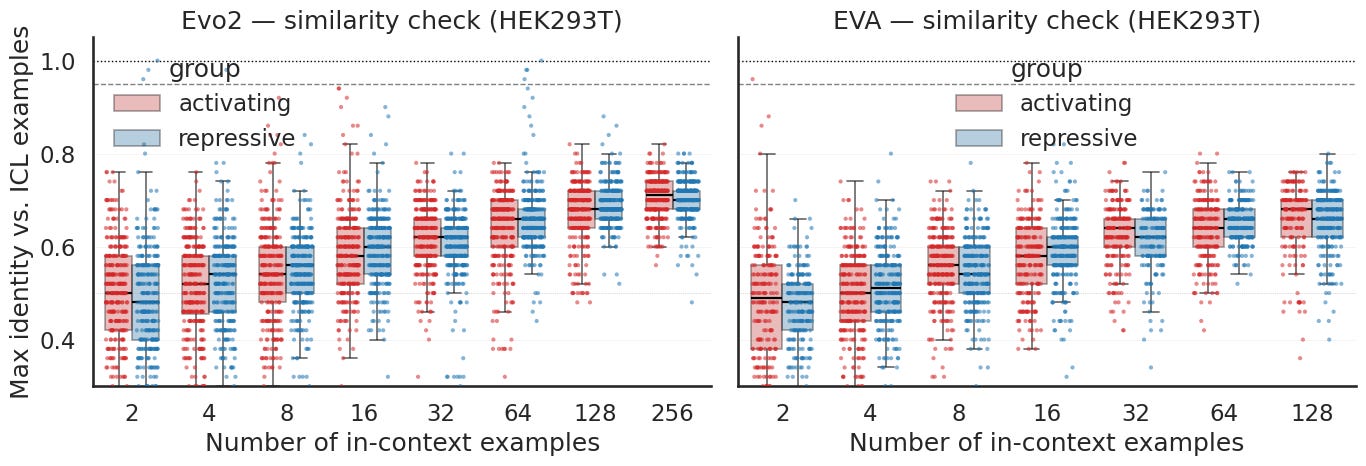
Identity does rise as you add examples, which makes sense, since a longer prompt gives the model more to lean on and more chances at a partial match. But it rises slowly and flattens out well short of copying. Even at the largest prompts a typical generation overlaps its nearest example by only around two thirds, and the bulk stay clear of the copy line at the top of the plot. This holds for both models and both prompt groups. So the sequences are genuinely new.
Why I find this interesting
One, this is in context learning at the sequence level, not the language level. The token here is a nucleotide. The pattern the model latches onto is not English or code but presumably some combination of k-mer composition, motif content, and structural propensity. Whatever that combination is, it transfers to a separate model trained on a separate readout. The two models have never spoken to each other and still agree, and the degree of agreement differed between them, most visibly on the harder of the two outputs.
Two, no training. No fine tuning. No reward model. No labeled dataset on the foundation model side. The cost of getting a usable generator of “high activity 5’ UTRs for this cell line” was the cost of sampling a few examples and gluing them with pipes. If the result holds up under bench validation, that is an efficient way to get there.
Three, this kind of prompt-based steering opens up a different mode of how we use foundation models internally. The path so far has been: collect data, train a specialist (T-Grammar), use the specialist to score or generate. The alternative path this experiment hints at is: collect data, use the specialist as an oracle to filter generations from a foundation model that is being steered by a small number of in context examples. We keep the specialist where it is most useful, predicting activity from sequence with proper calibration, and let the foundation model write plausible nucleotide sequences, without teaching it anything new. The comparison gives an early read on which model recovers the signal more reliably for this task.
One caveat here, we are validating in silico, with one model (T-Grammar) judging the other two (Evo2 and EVA). The interesting next step is the wet lab. If the Lab in the Loop generates a small set of prompt steered sequences, splits them into activating primed and repressive primed batches, and runs them through the same MPRA assay that produced the training data in the first place, the in context signal either survives in cells or it does not. That is the result that ultimately matters, and it is the one that would tell us whether the difference we see here is real or an artifact of one model judging another.
The promise of in-context learning has been around for a few years on the language side. Show a model a few examples, get a model that does the task. Seeing it on RNA sequence design, with two different foundation models agreeing on what counts as a good generation, was a striking thing to watch.