

The Case for Foundation Models in Biology
Rethinking scale: context, diversity, and design for biological foundation models

The past few years have seen biology embrace the ideas of large language models. Genomic and transcriptomic data, at some level, are also sequences with their own language and grammar. The cell often translates one to the other seamlessly. Why not then apply the same tricks that worked so well in language? Build massive models, train them with self-supervised objectives, and expect breakthroughs.
Reality has been more complicated. Across benchmarks, far smaller supervised models often outperform large pretrained ones. For some, this has led to the conclusion that scaling has failed in biology (or at least to a questioning of the underlying premise).
I think that conclusion is premature. Scaling is alive, but it plays out differently in biology than in text or images. Success depends on scaling the right axes — not just parameters, but context length, data diversity, objectives, tokenization, and architecture. And it depends on evaluating models fairly, with methods that reveal their strengths rather than obscure them.
In this post, I am laying out why scaling still matters for biology, why in fact foundation models are crucial for bringing biology into the next decade, and more importantly what dimensions we should be scaling, and how to think about evaluation.
Supervised Models Excel with Abundant Data, but Biology Is Far More Complex
In genomics, supervised models excel where labels are plentiful. Human and mouse genomes have benefited from massive data-generation consortia. Disease genetics, ENCODE-like catalogs of regulatory activity, popular immortalized cell lines; these are rich ecosystems where supervised models can thrive. Feed them thousands of transcriptomic and epigenomic tracks and they deliver excellent performance.
But biology extends far beyond this tight circle. Step into zebrafish, plants, or microbial consortia, and labeled data quickly thins out. Assays become noisy, annotations sparse, and sample sizes small. Training high-capacity supervised models is simply not feasible in these settings.
This is where foundation models matter. Pretraining across species and contexts lowers the barrier in data-poor domains. A model like Evo 2, trained on thousands of genomes across the tree of life, carries useful priors even for organisms studied by only a handful of labs. Its broad evolutionary grounding allows transfer: patterns learned from one species help interpret another. And remarkably, Evo 2 achieves state-of-the-art performance on both non-coding and coding variant prediction in humans despite being trained only on reference genomes, not on expensive omics tracks.

The lesson: supervised models may be dominating in rich-data niches for the time being. However, foundation models are indispensable in sparse ones. And it should be clear to everyone that much of biology is data-sparse.
Benchmarking Pitfalls: Why Evaluation Choices Matter
I feel like there is a tendency in our field to downplay the value of foundation models. But in reality, benchmarking of models requires care and attention. When foundation models are benchmarked, the results often hinge on methodological details that are easy to overlook.
1. Layer Choice Is Not a Nitpick
Transformers do not distribute information evenly across layers. Early layers capture local features; middle layers often encode rich structural and functional signals; late layers tilt toward autoregressive objectives. Picking the wrong layer to probe can make a model look weak.
Yet many benchmarks report results from a single layer or from a naive averaging of token embeddings. That choice can flip conclusions. For Evo 2, for example, the authors show that probing layer 20 yields excellent classification accuracy on BRCA1 variants; whereas, probing layers 1-3 produces far worse results. Without systematic layer sweeps and thoughtful pooling strategies, comparisons between models become unreliable.

2. Cropping Results Misses the Full Story
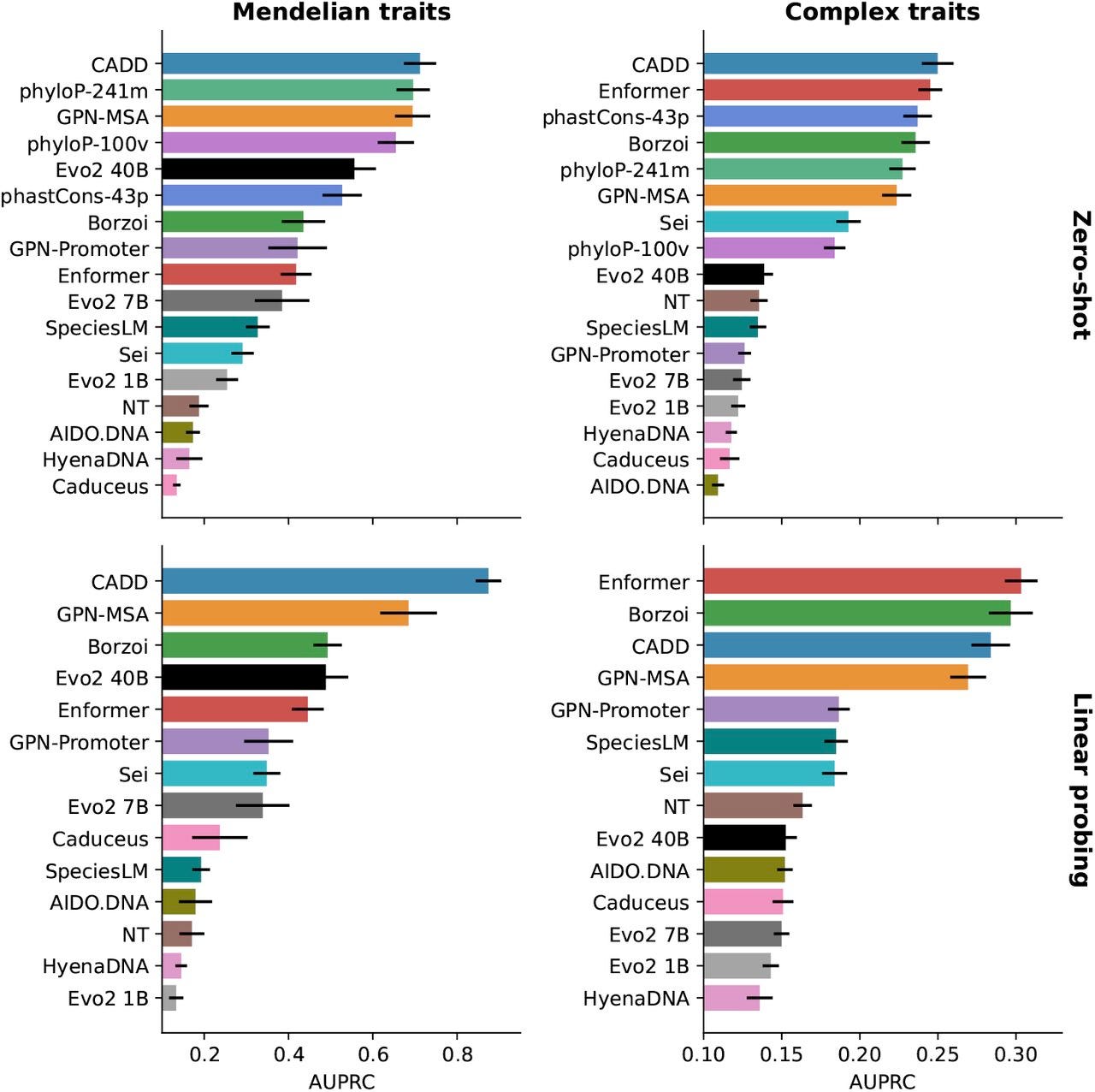
Benchmarks like TraitGym illustrate another problem. Evo 2 lags behind specialized models on some complex traits but outperforms them on Mendelian ones. This makes biological sense: Mendelian traits are often driven by strong-effect variants in coding or conserved regulatory regions, where evolutionary pretraining is powerful. Complex traits, by contrast, are polygenic and data-rich, favoring supervised models tuned to phenotype-specific signals.
But over and over again, critics simply share a cropped version of this evaluation. Sharing only the right-hand panel of a benchmark, the slice where supervised models win, gives a skewed impression. Reporting full grids reveals the trade-offs: foundation models recognize evolutionary disruption; supervised models capture subtle statistical associations. Even in humans where we have plenty of data, both stories are true, and both matter.
Noise Today Can Be Structure Tomorrow
One common critique is that most of the genome is “noise.” Intergenic regions make up ~75% of mammalian genomes, and many positions seem unconserved. If only ~10% of nucleotides are conserved across species, the argument goes, why waste model capacity predicting the rest?
This view misrepresents regulatory biology. Intergenic regions are not uniform deserts. They are patchworks of functional elements embedded in less constrained sequence. Enhancers, silencers, and insulators sit scattered across these expanses, looping over vast distances to control genes. Chromatin domains partition the genome into 3D compartments that matter for transcriptional regulation.
The ENCODE 2025 encyclopedia catalogs millions of candidate regulatory elements in human and mouse alone. Many of these lie in “non-conserved” territory. Sequence-level conservation is not the right proxy: regulatory function often persists through network-level conservation, where different motifs evolve to achieve the same control logic.
When models struggle to learn from intergenic regions, it does not mean those regions are meaningless. It means our objectives are insufficient. Absent large swaths of data, standard next-token prediction may miss distal and sparse dependencies. Here, addition of supervised tasks, predicting chromatin contacts, enhancer-promoter loops, or expression changes under perturbation, could teach models to extract real structure from apparent noise.
Scaling Beyond Parameters
In public discourse, scaling is often shorthand for “adding parameters.” Bigger is better. But in biology, parameters are only one axis, and sometimes not even the most important one.
Context length. Many regulatory interactions span hundreds of kilobases. Standard autoregressive training with limited windows will miss them. Long-context models like Evo 2 and AlphaGenome, with million-token windows, show why context scaling can be more impactful than sheer size.
Tokenization. Single nucleotide tokens are simple but not necessarily optimal. Codons, kmers, or adaptive schemes can encode biological structure better, letting smaller models outperform larger but poorly tokenized ones.
Readouts. Linear probes on the right intermediate layer can beat more complex architectures on the wrong one. Benchmarking must separate engineering choices from fundamental model limits.
Data breadth. A model trained only on human data will struggle with plants. One trained only on coding sequences will miss regulatory grammar. Scaling species diversity, assay types, and experimental contexts often delivers larger gains than adding layers.
Scaling in biology is multi-dimensional. The critical question is not “how big is the model?”, but “what axes are we scaling, and are they aligned with biological signals”?
Architectures for Biology, Not Just Borrowed from Language
Language models advanced because architecture and data evolved together. Self-attention unlocked long-range dependencies; tokenization improved; datasets scaled; instruction tuning taught models to use their knowledge.
Biology will follow a similar trajectory, but with its own twists. The signals we care about are inherently bidirectional, multimodal, and structured in 3D.
Bidirectionality. Enhancers regulate promoters upstream and downstream. RNA folding depends on base-pairing across both directions. Autoregressive models that only look backward miss half the story.
Long-range, sparse interactions. Unlike text, where nearby words matter most, biological regulation often skips over large spans. Sparse attention or hierarchical models may capture this better.
Multi-scale patterns. Chromatin domains, local motifs, 3D genome structure; biology is layered in ways language is not. Standard transformers may not capture these efficiently.
We likely have not yet found the optimal architecture for biological sequences. Borrowing directly from NLP will take us part of the way, but breakthroughs will come from designs tailored to biological invariances.
The Data Frontier: Diversity, Perturbations, and Synthetic Biology
In our field, there is also a sense that we are approaching the limits of meaningful biological data. This is simply wrong. OpenGenome2 and similar collections are milestones, not endpoints. Sequence diversity is vast; microbial communities, metagenomes, plants, and understudied clades remain under-sampled. Functional diversity is even larger. Perturbation assays, multi-modal single-cell data, and time-series measurements are only beginning to scale.
But most importantly, we are no longer limited to natural data. Genome-scale generative processes and genome foundries mean that we can design synthetic sequences and test them at scale, effectively creating new training data beyond what evolution provides. Evo itself demonstrates this: models that generate and evaluate synthetic variation expand the dataset in directions natural diversity never explored.
Data growth in biology is not about chasing the trillion-token thresholds of NLP. It is about expanding along the biologically relevant axes: species, conditions, modalities, perturbations, and designed diversity.
Foundation Modeling Is Needed More Than Ever
Taken together, these points argue for a reframing. Scaling in biology is not dead. It is conditional.
Foundation modeling helps most where data is sparse.
Scale fails if we probe the wrong layers or delude ourselves by cropping benchmarks.
Scale needs objectives that respect biological regulation.
Scale is multi-dimensional: context length, tokenization, data breadth, architecture, not just parameters.
Scale depends on data diversity, including synthetic generation.
When these conditions are met, scaling delivers. Evo 2’s performance on variant interpretation, foundation models’ ability to transfer across species, and the early success of long-context architectures are all evidence. The story is not failure; it is refinement.
Conclusion
Scaling has always been about more than size. In biology, it is about matching objectives, architectures, and data to the underlying signals. Dismissing scaling because supervised models win in data-rich niches misses the broader picture.
The right framing is this: scaling is alive, but it must be given the right instructions, such as longer contexts, better tokenization, richer objectives, broader data. Under those conditions, scaling unlocks biological insight in places where no supervised model could even start.
Scaling works. We just need to scale the things that matter.